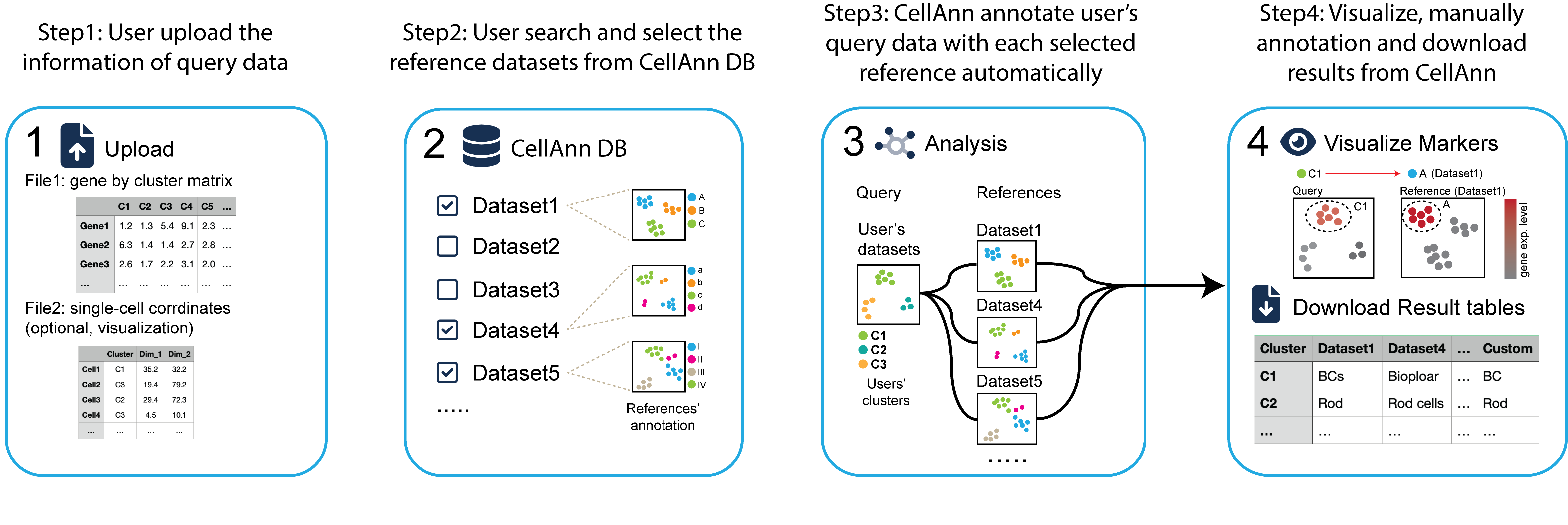
CellAnn is a web application for predicting cell types of single-cell clusters based on published reference datasets. CellAnn provides a comprehensive scRNA-seq reference database and users can easily find the relevant reference datasets in their analysis.
What's new:
2023-7-5 update Updated datasets. 2023-3-5 update Added Installation instructions for running CellAnn in local computer. 2023-3-1 update Added more datasets for Mouse blood. 2023-2-26update Added bars to adjust the ranges of gene expression in Step4. 2023-2-23bugfix Now users can stay on the website without losing their connection for up to 1 hour. 2023-1-17update CellAnn version1.0 is online!
Database:
Human:
Mouse:
Workflow:
Citation:
Pin Lyu, Yijie Zhai et al. CellAnn: A comprehensive, super-fast, and user-friendly single-cell annotation web server[J]. bioRxiv.
Analysis
There are 4 major steps running cell type alignment:
Step1:Upload your average expression files
Step2:Select datasets from CellAlign database
Step3:Mapping your cluster to your selected datasets
Step4*:Visualize your results and generate your custom cell type annotation
We provide example inputs generated from different studies:
1.Download example inputs from the Mouse Tabula Muris study
here
.
2.Download example inputs from the Human Pancreas studies
here
.
3.Download example inputs from the Human PBMC study
here
.
Frequently Asked Questions:
Q1: Before use CellAnn, how to prepare the input files?
for R seurat users:
1.Install devtools (for installing GitHub packages) if it isn't already installed.
### install devtools in R ###
if (!requireNamespace("devtools", quietly = TRUE)) install.packages("devtools")
2.Prepare your Seurat object with “prepare_CellAnn” function.
### download scripts ###
devtools::source_url("https://raw.githubusercontent.com/Pinlyu3/CellAnn/main/prepare_CellAnn.R")
### prepare_CellAnn function:
### parameter: seurat_obj: your seurat obj
### parameter: folder(character): CellAnn ouput files will be output to this path, default is your current folder
### parameter: sample_name(character): names of this sample, such as "Liver_1", "eye2_2" etc.
### parameter: matrix_name(character): default will use the "RNA" counts matrix in your seurat object
### parameter: dims(character): "umap" or "tsne". default is 'umap'
### parameter: cluster(character): the name of the column which stored cluster information in the metadata of your Seurat object. default is 'seurat_clusters'
prepare_CellAnn(seurat_obj,folder=folder,sample_name='your_samples_name',matrix_name='RNA',dims='umap',cluster='seurat_clusters')
### After run prepare_CellAnn function, you will find 2 prepared files under your folder
list.files(folder)
for Python scanpy users:
### load library and your query datasets:
import scanpy as sc
import pandas as pd
adata = sc.read('data.h5ad')
### Calculate the average gene expression within each cluster
### we assume that the raw counts are stored in adata.raw.X
### the cluster information stored in adata.obs['cluster']:
### the gene_names stored in adata.var_names
### the cell name stored in adata.obs_names
### the 2 dimension UMAPs strored in adata.obsm['X_umap']
### parameter: sample_name(character): names of this sample, such as "Liver_1"
def cluster_average(adata,sample_name):
cluster_df = adata.obs['cluster']
gene_names = adata.var_names
cell_names = adata.obs_names
mat_df=pd.DataFrame.sparse.from_spmatrix(adata.raw.X)
####
row_annotations = np.array(cluster_df)
unique_annotations = np.unique(row_annotations)
####
merged_matrix = np.zeros((len(unique_annotations), mat_df.shape[1]))
####
mat_df_mat = np.array(mat_df)
for i, annotation in enumerate(unique_annotations):
###
rows_to_sum = (row_annotations == annotation)
rows_to_sum_mat = mat_df_mat[rows_to_sum,:]
###
sum_rows = np.sum(rows_to_sum_mat, axis=0)
sum_rows_norm = sum_rows / np.sum(sum_rows) * 1e5
sum_rows_log = np.log(sum_rows_norm+1)
###
merged_matrix[i, :] = sum_rows_log
#### output to csv #####
df_log = pd.DataFrame(merged_matrix.T)
df_log.columns = unique_annotations
df_log.index = gene_names
df_log.insert(0,"GENE",df_log.index)
####
df_log = df_log.round(3)
Output_Step1_name = sample_name + '_CellAnn_Step1_input.txt'
df_log.to_csv(Output_Step1_name, index=False,sep='\t')
####
df_umap = pd.DataFrame(adata.obsm['X_umap'])
df_umap.columns = ["dim1","dim2"]
df_umap.insert(0,"cell",cell_names)
df_umap.insert(1,"cluster",row_annotations)
Output_Step4_name = sample_name + '_CellAnn_Step4_input.txt'
df_umap.to_csv(Output_Step4_name, index=False,sep='\t')
### run cluster_average with your adata to get the input of step1 and step4:
cluster_average(adata,sample_name="your_sample_name")
Q2: How to use the CellAnn ?
Q3: How to add the CellAnn results to Seurat ?
1.You can integrate cellann outputs to seurat object in R with the function “merge_CellAnn_Seurat”.
2.Make sure you have installed “readr” library.
### download scripts ###
devtools::source_url("https://raw.githubusercontent.com/Pinlyu3/CellAnn/main/merge_CellAnn_Seurat.R")
### merge_CellAnn_Seurat function:
### parameter: seurat_obj: your seurat obj
### parameter: cluster(character): the name of the column which stored cluster information in the metadata of your Seurat object. default is 'seurat_clusters'
### parameter: cell_anno: the annotation file from CellAnn Step3 or Step4
Your_new_seurat <- merge_CellAnn_Seurat(seurat_obj=Your_old_seurat,cluster='seurat_clusters',cell_anno='step3orstep4_output.txt')
### After run merge_CellAnn_Seurat function, you will find the cell_anno has been merged into your seurat
head([email protected])
Q4: I cann't find my interested datasets in CellAnn.
Please contact us and provide the link (e.g. GEO number) to your datasets and related papers.
Issues using CellAnn?
CellAnn is currently in beta. If you find a bug, please report an issue on Github
with the Bug Report form.